
유전자 편집이란?
유전자 편집은 살아있는 유기체의 Genomic DNA를 결실, 삽입, 대체 또는 변형하는 유전자 조작입니다. 유전자 편집은 다양한 기술을 통해 DNA를 손상시키기 위한 영역 특이적 표적화이며 항상 복구 기전을 포함하는 것은 아닙니다. 비활성화와 교정의 두 가지 기술로 구성됩니다.
비활성화는 표적 유전자의 전환을 포함하며, 교정은 유전자를 손상시켜 결함이 있는 유전자의 복구를 용이하게 합니다. 유전자 편집은 신약 개발, 유전자 수술, 동물 모델, 질병 연구와 치료, 식품, 바이오 연료, 생체 재료 합성 등 수많은 분야에서 엄청난 잠재력을 가지고 있습니다.
최근에는 주요 유전자 편집 기술인 CRISPR가 널리 사용되었지만, 유전자 편집은 1900년대 후반에 처음 연구되었습니다. 과거에는 야심찬 응용 분야였던 CRISPR가 시작된 이후로, 유전자 치료는 유전자 편집의 가장 인기 있는 응용 분야가 되었습니다. 이는 기존의 유전 물질에 유전자를 추가하여 불완전 유전자나 누락된 유전자를 보충하는 유전자 보강과 질병 관련 DNA를 직접 변형하여 질병을 치료하는 유전자 편집의 두 가지 접근 방식을 통해 달성할 수 있습니다.
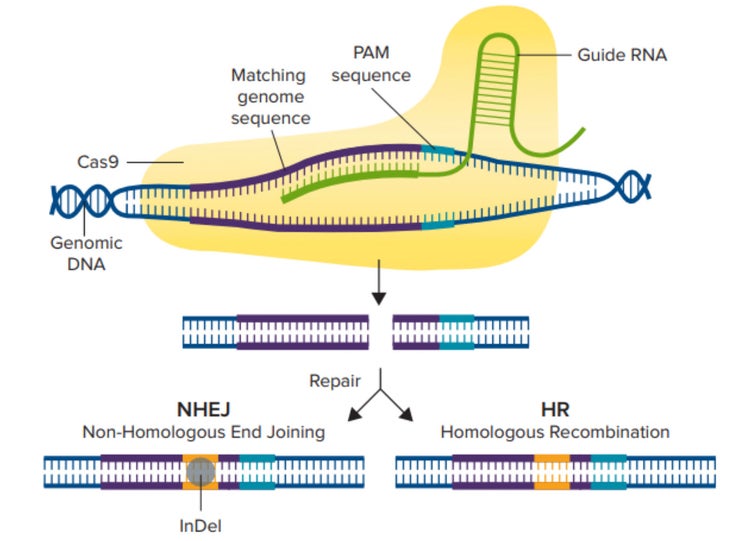
CRISPR/Cas9 기전 Cas9 효소는 먼저 가이드 RNA에 결합한 다음 3-뉴클레오티드 PAM 염기서열 바로 앞에 있는 이에 대응하는 Genomic 서열에 결합하여 활성화됩니다. 그런 다음 Cas9 효소는 이중 가닥을 절단하고 NHEJ 또는 HDR 경로를 사용하여 DNA를 복구하여 편집된 유전자 염기서열을 생성합니다.
crRNA와 유사한 가이드 RNA(gRNA)는 유전자의 특정 영역을 표적으로 하도록 설계되었으며 Cas9 효소는 숙주 세포의 유전체 특정 영역에서 이중 가닥을 절단할 수 있습니다(그림 1). 이중 가닥이 절단된 후 세포에서는 두 가지 복구 경로인 비상동 말단 연결(NHEJ) 경로 또는 상동성 지향 재조합(HDR) 경로 중 하나가 발생합니다. NHEJ 경로는 일반적으로 염기 삽입 또는 결실(indel)을 통해 유전자를 파괴하는 데 사용되는 반면, HDR 경로는 2개의 유사하거나 동일한 DNA 분자 간 염기서열을 교환함으로써 리포터 유전자 또는 편집된 염기서열의 Knock-In에 사용할 수 있습니다.
CRISPR 공학을 사용한 유전자 편집 확장
‘CRISPR(Clustered Regularly Interspaced Short Palindromic Repeats)’ – 일정한 간격을 두고 주기적으로 분포하는 짧은 회문 반복서열 이러한 DNA 염기서열은 박테리아, 고세균과 같은 원핵생물에서 면역계의 일부로 처음 발견되었으며, 2012년 이후 유전자 편집 도구로서 중요성을 갖게 되었습니다(Jinek 외, 2012). 이는 농업, 질병 모델링, 유전자 치료, 신약 개발 등 수많은 응용 분야에서 매우 유망합니다. 정밀도가 높기 때문에 삽입(Knock-In), 결실(Knockout), 기타 DNA 염기서열 변형을 위한 완벽한 도구입니다. 이는 TALENS, ZFNS와 같은 기존의 진부하며 고비용의 유전자 편집 도구를 상당 부분 대체했습니다.
CRISPR 염기서열에는 각 회문 반복서열 뒤에 스페이서라고 하는 이전 침입자 바이러스의 DNA가 포함되어 있으며 이러한 DNA는 향후 유사한 바이러스를 탐지하고 파괴하는 데 도움이 됩니다. 이 기전(Jinek 외, 2012)을 이해하여 진핵세포에서 CRISPR의 첫 번째 사용(Cong, L, 외, 2013)이 이루어졌고 이후 다른 세포 유형과 다른 분야에 속하는 유기체에 사용되었습니다. CRISPR – Cas9 시스템에는 리보핵단백질 복합체를 형성하는 두 가지 주요 구성요소가 있습니다. 첫 번째 구성 요소 또는 가이드 RNA는 유전체의 상보적 DNA 염기서열에 결합하고, Streptococcus Pyogenes(SpCas9)의 두 번째 구성 요소 Cas9은 표적 부위에서 이중 가닥을 절단합니다. Protospacer Adjacent Motif(PAM)는 업스트림에서 절단이 일어나도록 핵산분해효소가 초기에 결합하는 영역입니다. 서로 다른 CRISPR 핵산분해효소에는 서로 다른 PAM 영역이 존재하며 일단 절단이 이루어지면 세포 복구 시스템이 활성화되고 유전체에 대한 편집도 시작됩니다.
유전자 편집 실험과정
확인된 편집 세포주를 얻기 위해 CRISPR 기전을 사용하는 유전자 편집 실험과정에는 다양한 단계가 있습니다. 올바른 도구를 사용하여 이러한 단계를 효과적으로 최적화하면 다양한 과학적 발전의 시간, 노력, 비용을 줄이는 효율적인 과정에 기여합니다. 이 접근 방식은 R&D를 가속화하고, 신약 개발, 질병 치료, 유전자 변형 작물 생산 등의 혁신을 돕습니다. 우리는 전 세계 과학 공동체가 유전자 편집을 통해 노력의 결실을 얻을 수 있도록 지원하고자 관련된 단계와 효과적인 솔루션에 대해 논의합니다.
CRISPR/Cas9 유전자 편집 Validation을 위한 연구 솔루션
Molecular Devices의 장비 제품군은 유전자 편집 노력의 성공을 보장하는 실험을 수행/Screening하는 데 효과적으로 사용할 수 있습니다. 새로운 CloneSelect Imager Florescence(CSI-FL)는 단일 세포 프린팅, Transfection 효율성, Cell Confluency, 다중 채널 형광 Screening 일자 이후 0일차에 Monoclonality를 보장하며 더 짧은 추적 시간, 낮은 과대 계대배양 위험, 로봇 공학을 통해 유전자 편집 유효성을 Validation합니다.
또한, 우리의 SpectraMax i3x Multi-Mode Microplate Reader를 사용하여 Transfection 효율을 평가하고 Cell Growth를 Monitoring하고 DNA 및 단백질을 정량분석하고 ScanLater Western Blot 분석을 통해 CRISPR/Cas9 편집을 Validation할 수 있습니다. 자가포식소체의 고품질 이미지는 ImageXpress Micro Confocal System을 사용하여 얻을 수 있으며 MetaXpress HCI 소프트웨어로 모든 세포에서 개별 자가포식소체를 식별하고 정량분석하여 CRISPR/Cas9 유전자 편집에서 발생하는 표현형 변화를 분석할 수 있습니다.